- Introduction
The aim of this study was to determine the crystalline structure of Aspirine, to establish its solid state molecular geometry and to generate a simulated X-ray powder diffraction pattern corresponding to the structure, which could be compared with experimental patterns.
- X-ray experimental parameters
Slow evaporation from Recristallisé dans un
mélange eau-éthanol affords crystals suitable for X-rays diffraction studdies.
A single crystal selected by observation under a binocular microscope was mounted on the goniometric head of a KappaCCD diffractometer.
Intensities were collected at room temperature (T=293 K), with the use of a graphite monochromated Cu Kα radiation ( λ = 1.54178 Å ).
Systematic investigation of the diffraction nodes indicate that the crystal belong to the monoclinic system, with a primitive Bravais lattice. The unit cell parameters are:
a (Å) = 11.43 ; b (Å) = 6.59 ; c (Å) = 11.40 ; α (°) = 90.01 ; β (°) = 95.68 ; γ (°) = 89.98
In view of the number of atoms in the Aspirine molecule and of the unit cell volume, it is concluded that this unit cell must contain 4 molecules having the formula
C9 H8 O4
which is equivalent to a calculated density of 1.401. The number of reflections collected was 1613, of which 1527 were unique.
Determination of the space group was achieved unequivocally due to the presence of a systematic extinction along the monoclinic axis together with a systematic zonal extinction othogonal to this axis.
- Structure Refinement
The structure was solved by a direct methods using the SIR software [1]; and refined on F² by full least squares methods with SHELXTL [2]. All non hydrogen atoms were refined with anisotropic displacement parameters, a riding model was used for hydrogen atoms. Final agreement values are R1 = 0.0400 (observed reflections) and wR2 = 0.1404 (all data) for 1527 and 128 parameters, with a goodness of fit of 2.004.
-
Description of the structure
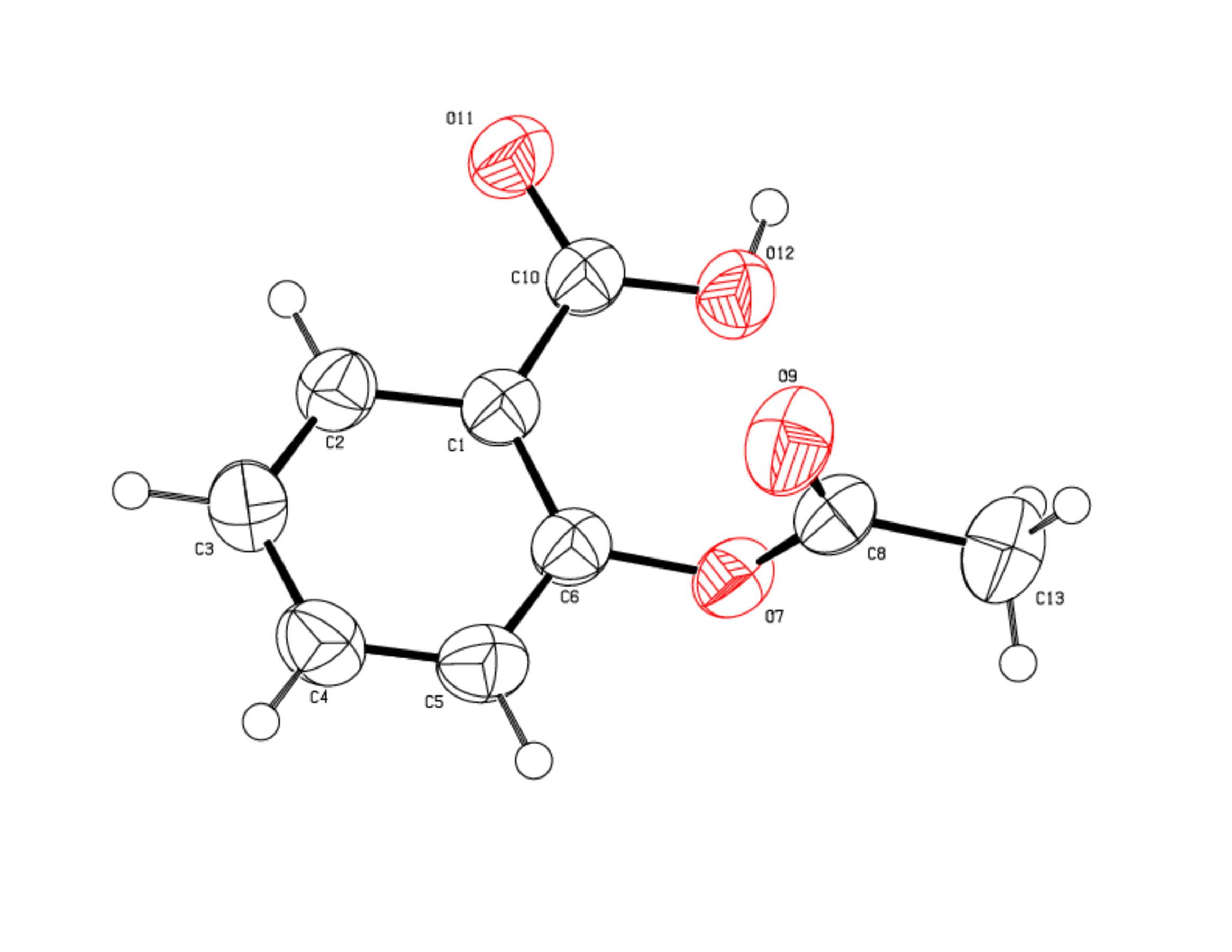
Crystal data, X-rays experimental parameters and structure refinements are given in Table 1. Table 2 lists the positional parameters for all independent non-hydrogen atoms together with their equivalent isotropic displacement parameters. Bond lengths and angles are listed Table 3 and 4. Hydrogen positions are reported Table 5.
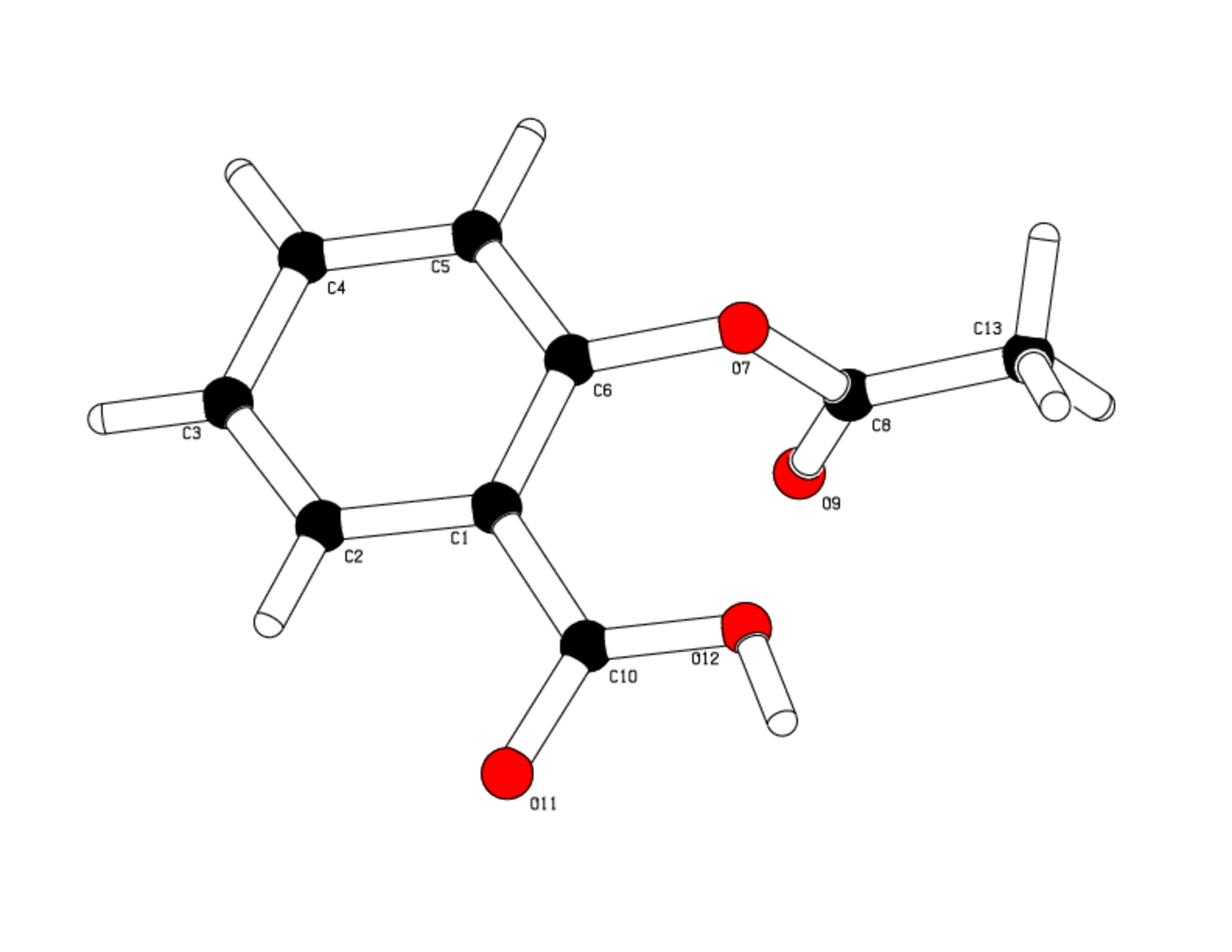
The compound (figure 1) crystallize in the space group P 21/c, the asymmetric unit of the crystal is made up of 1 molecule of Aspirine, thus 4 formula are present in the unit cell.
No additional molecule like organic or water is found.
Examination of the molecular structure confirms that all bond angles and lengths stand in the standard range values.
-
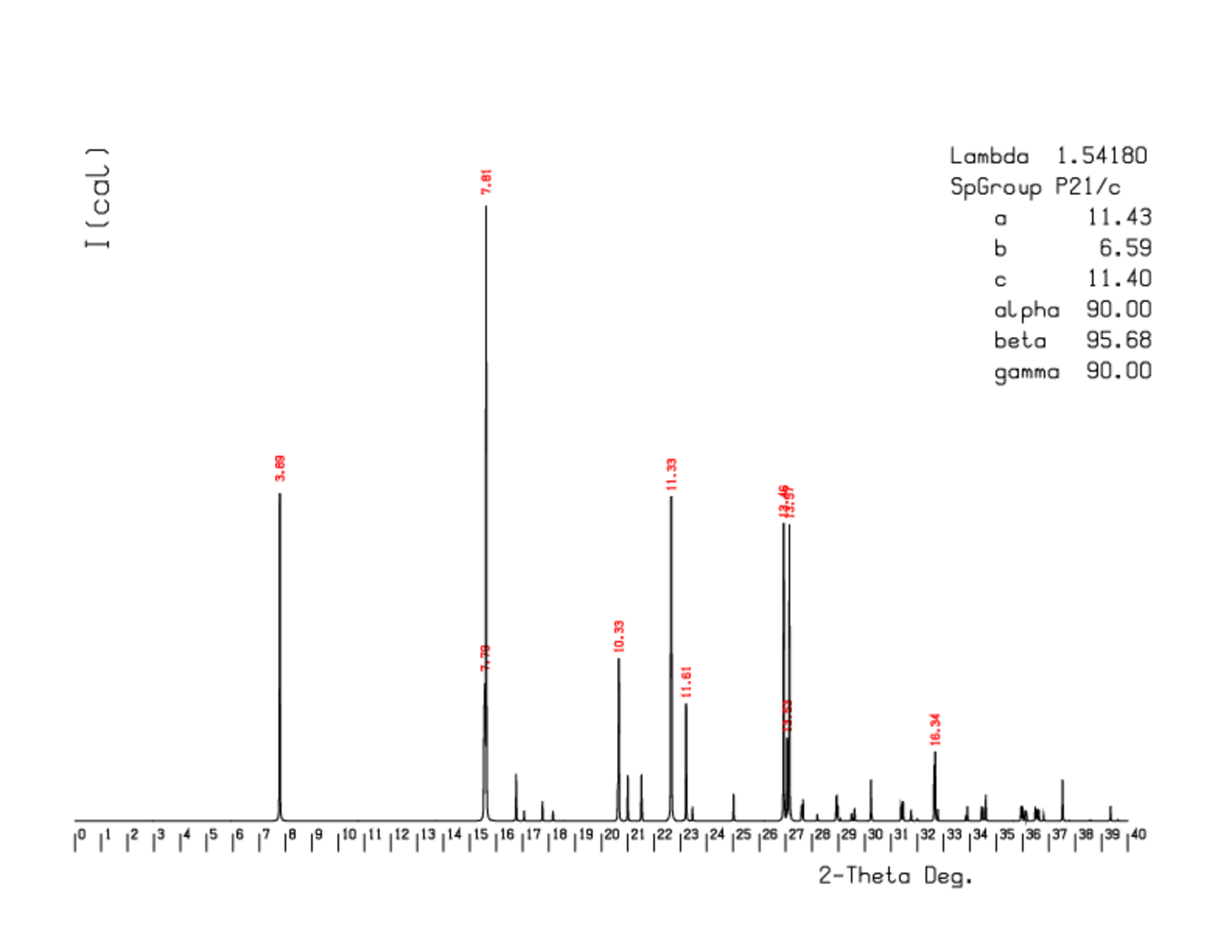
Comparison of an experimental diffraction pattern with a simulated diffraction pattern.
A simulated diffraction pattern (schema 3) was produced from the experimentally determined crystalline structure. A powder diffraction pattern can be compared to this theoretical pattern to demonstrate the nature of the crystalline structure. Minor differences can be explained by preferential orientations in the powder.
-
Conclusion
The crystalline structure of Aspirine was determined by X-ray diffraction on a single crystal, allowing the generation of a reference powder pattern.
-
References
[1] Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Burla, M. C.; Polidori, G.; Cavalli, A. J. Appl Crystallogr. 1994, 27, p 435-436.
[2] Sheldrick, G. M. SHELXTL-Plus, Rel. 5.03; Siemens Analytical X-ray Instruments Inc.: Madison, WI, 1995.